Computational Photochemistry Study of 2-Hydroxyazobenzene
Institute of Advanced Computational Sciences, Stony Brook University | Advisors: Benjamin G. Levine, Arshad Mehmood
The molecule 2-hydroxyazobenzene is the building block for many dyes and has the potential to undergo excited-state intramolecular proton transfers (ESIPT). While its dominant non-radiative relaxation pathway has been studied using static multi-reference methods, there is a lack of research on its excited state molecular dynamics using ab initio methods. Our work aims to address this gap by investigating the excited state dynamics using the ab initio TAB method developed by the Levine Research Group. This research will enhance our understanding of the photochemistry of 2-hydroxyazobenzene, providing a foundation for further investigations of similar systems involving multiple ESIPT reactions.
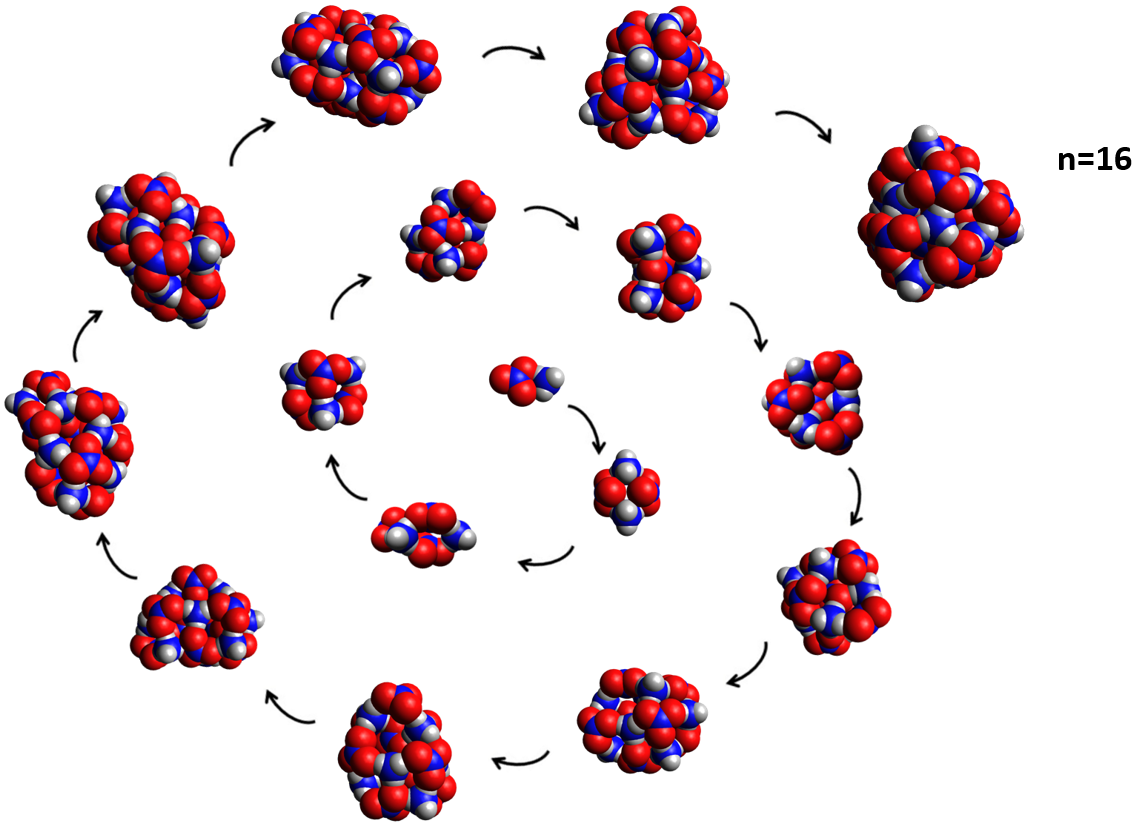
Decomposition and Growth of Ammonium Nitrate Clusters
Department of Chemistry, The Cooper Union for the Advancement of Science and Art | Advisors: Robert Q. Topper
A better understanding of the formation and decomposition of aerosolized ammonium nitrate species should lead to improvements in the modeling of aerosol haze formation. We found that simulated annealing Monte Carlo optimizations via TransRot combined with a well-chosen density functional theory model can be used reliably to predict minimum-energy structures and interaction energies for the cation and anion clusters observed in mass spectra as well as for neutral nanoparticles. Our calculations made it possible to rationalize the pattern of peaks in the mass spectra through computation of the binding energies of clusters involved in various growth and dissociation pathways. We were the first to identify the structures corresponding to all detectable species in both the positive and negative ion mass spectra of ammonium nitrate. We also study the structures and interaction energies of larger (NH4NO3)n nanoparticles (shown to the right).